Amyotrophic Lateral Sclerosis – TDP-43 Transgenic Mouse Model
Target the pathological hallmark of Frontotemporal Dementia (FTD) and almost all Amyotrophic lateral sclerosis (ALS) patients using the TDP-43 Transgenic Mouse Model
TDP-43 Transgenic Mouse Model Key Characteristics
The TDP-43(Q331K) transgenic mouse model represents a well-characterized (Arnold et al., 2013; Watkins et al., 2012) and relevant preclinical research tool to test efficacy of novel therapeutics targeted against amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). TDP-43 pathology is observed in approximately 95-97% of ALS cases and in ~50% of FTD cases, making TDP-43 a major therapeutic target across the ALS-FTD disease spectrum. The TDP-43(Q331K) mouse model carries an ALS-associated Q331K mutation (Sreedharan et al., 2008) in the C-terminal domain of human TDP-43 directed to brain and spinal cord by the mouse prion protein promoter, a region critical for protein regulation and RNA binding.
This single transgenic TDP-43 mouse model of ALS and FTD has been shown in literature (Arnold et al., 20123; Watkins et al., 2021) and confirmed by InnoSer’s validation experiments (link to figures here) to recapitulate the pathophysiological disease features of human TDP-43 proteinopathy. Although this model shows cytoplasmic TDP-43 accumulation (see figure here) with insoluble cytoplasmic TDP-43 aggregates appearing only at 24 months of age in both cortex and spinal cord (Mitchell et al.,2015), motor neuron disease features of human TDP-43 proteinopathy in this mouse model develop starting at 4 months of age without robust cytoplasmic TDP-43 aggregation and nuclear clearing (Arnold et al., 2013; Wakins et al., 2012; Mitchell et al., 2015). In line, this single transgenic TDP-43 mouse model avoids artefacts (such as premature lethality) associated with other TDP-43 mouse models featuring high TDP-43 overexpression leading to insoluble cytoplasmic TDP-43 inclusions and TDP-43 nuclear clearing (such as observed in the TDP-43 TAR4/4 mouse model; Wils et al., 2010) or the double transgenic TDP-43 mouse model described by Mitchell et al., 2015), allowing you to perform longitudinal motor function assessment studies focusing on demonstrating efficacy on the functional level of ALS and/or FTD.
✓ InnoSer’s team has extensively validated the TDP-43Q331K model from 4 weeks of age, demonstrating early, progressive motor deficits that become robust and reliably detectable by 4 months of age in line with literature (Arnold et al., 2013; Watkins et al., 2021; Mitchell et al., 2015)
✓ Motor function battery includes Rotarod, Balance beam, CatWalk gait analysis, Grip strength testing, weightlifting test, NeuroScore
✓ Translational readouts include sciatic nerve conduction studies (NCS; significant deficits observed from 7 weeks of age) and biomarker quantification of the neuronal injury ALS biomarker plasma NfL (significantly elevated from 5 weeks of age)

As part of InnoSer’s neurology ALS mouse model portfolio, we also offer efficacy studies in the SOD1-G93A transgenic ALS mouse model. However, as each model is unique, modelling distinct pathophysiological of ALS, we recommend you discuss the most appropriate model with our neurology study directors.
InnoSer’s neurology expert team possesses relevant experience in working with multiple therapy types, including small molecules, peptides, enzymes, oligonucleotides, gene therapy (viral vectors, e.g., AAVs), and immunotherapies (antibody/vaccine immunotherapies).
Your ALS Research Starts Here.
View study timelines, recommended readouts, and example data featuring behavioral across different ALS mouse models.

TDP-43 Transgenic Mouse Model Sample Data
The single transgenic TDP-43(Q331K) mouse model develops robust motor neuron disease pathophysiology features in the absence of overt cytoplasmic mislocalization and cytoplasmic TDP-43 aggregates/ inclusions and TDP-43 nuclear clearing, in line with literature (Arnold et al., 2013; Watkins et al., 2015; Mitchel et al., 2015) and other single transgenic TDP-43 mouse models such as the TAR6/6 TDP-43 mouse model (Wils et al., 2010; Scherz et al., 2018). Cytoplasmic aggregation of TDP-43 into insoluble inclusions in cortical and spinal neurons has been observed in this mouse model at 24 months of age (see supplementary figure 4 in Mitchel et al., 2015), making it more suitable for longitudinal studies examining rescue of motor function deficits in the setting of ALS and/or FTD.
InnoSer’s in-house validation of the TDP-43 mouse model included a qualitative assessment (performed in the same staining session and imaged under the same conditions) of TDP-43 (mouse and human TDP-43) cellular localisation, confirming an increase in the level of both cytoplasmic and nuclear TDP-43 compared to non-transgenic littermates. Note: Non-transgenic TDP-43 littermates show faint nuclear TDP-43 signal due to the presence of endogenous mouse TDP-43.

Motor function tests reveal early stage, and progressive disease development in female TDP-43Q331K mice
Motor function phenotyping was performed in two separate experiments in young (4-16 weeks of age; “early disease stage” phenotyping) and old (16-24 weeks of age; “late disease stage” phenotyping) female mice. Female hemizygous TDP-43Q331K mice exhibit progressive, reduced muscle strength assessed via the inverted grid test.
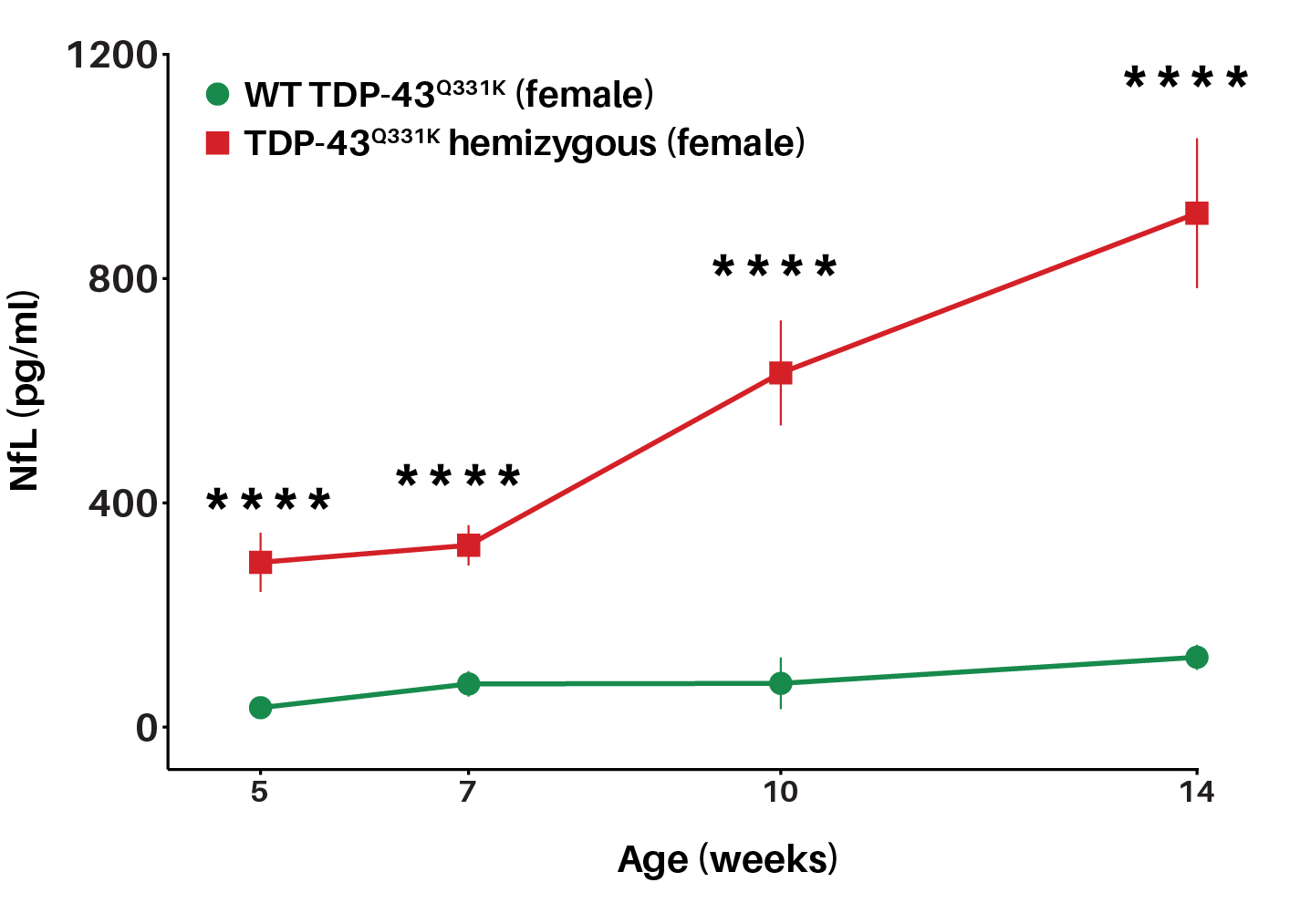
TDP-43Q331K mice show significant elevations in the neuronal injury biomarker, NfL, as early as 5 weeks of age
Neurofilament light chain (NfL) levels are significantly increased in TDP-43Q331K mice as early as 5 weeks of age. Longitudinal measurements of plasma NfL from 5 to 14 weeks of age show that female TDP-43Q331K mice exhibit significant elevations in plasma NfL compared to wild-type littermates, indicating early-onset and progressive neuronal injury phenotype. Plasma NfL was assessed via MSD (Kit number # K1517XR).
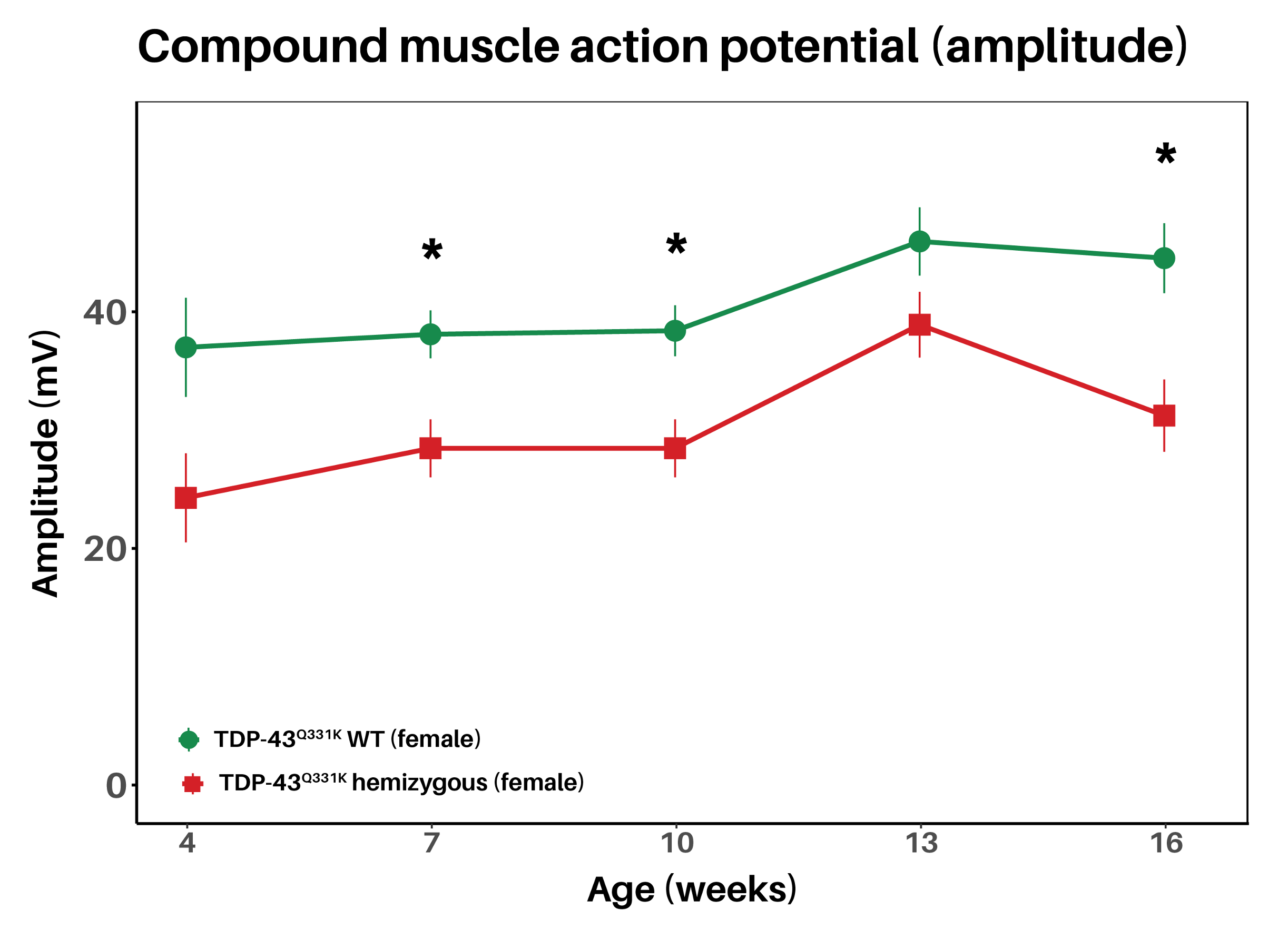
TDP-43Q331K mice show reduced CMAP amplitude from 7 weeks of age, before onset of robust motor function deficits indicating early motor unit dysfunction
Electrophysiological readouts reveal a reduction in compound muscle action potential (CMAP) amplitude in TDP-43Q331K mice from 7 weeks of age. Nerve conduction studies can be performed longitudinally in the same animal, enabling repeated measures of disease progression.
TDP-43 Transgenic Mouse Models Readouts
Biological Readouts
Test the efficacy of your treatments with the following biological readouts:
- Sciatic nerve conduction electrophysiology study (compound muscle action potential [CMAP] and nerve conduction velocity [NCV])
- Biomarker analysis (e.g.. plasma NfL)
The People Behind Your Research

Thomas Vogels, PhD, In Vivo Neurology Study Director
Leads an expert team of scientists with vast experience in our Neurology models to help you choose the right model and guide your optimal study design. We provide the solution to accelerating your drug development.
Struggling with getting your therapeutics across the blood-brain-barrier?
Discover how SonoCloud® ultrasound-mediated BBB disruption can improve brain uptake — without altering your compound.

Frequently Asked Questions
What is the TDP-43Q331K mouse model?
TDP-43Q331K model is a transgenic mouse model harbouring an ALS-associated mutation (Q331K) in human TARDBP gene. The TDP-43 model expresses mutant human TDP-43 (i.e., Q331K), a pathology present in ~95% of ALS and ~50% of frontotemporal dementia (FTD) cases, making this model highly relevant for preclinical efficacy testing of novel compounds not only for ALS, but also for ALS-FTD spectrum research.
This ALS mouse model was previously validated by Watkins et al., 2021, who demonstrated that TDP-43 mice display progressive motor deficits, supporting its use as a relevant tool for ALS and FTD preclinical research.
As a preclinical ALS CRO, InnoSer has characterized the disease progression of TDP-43Q331K mice from 4 weeks until 35 weeks of age (∼8 months) across multiple endpoints:
- Motor function readouts: Neurological scoring (NeuroScore), Rotarod, Inverted grid test, Weightlifting rest, gait deficits assessed via CatWalk, and behavioural deficits assessed via automated home-cages
- Electrophysiological readouts: nerve conduction studies including compound muscle action potential (CMAP)
- Biomarker readouts: plasma neurofilament light chain (NfL)
Has the TDP-43Q331K mouse model been used to test known ALS therapeutics or comparator compounds?
While the TDP‑43Q331K transgenic mouse model has been extensively characterized for progressive motor and neuromuscular deficits, published studies to date have not reported standard therapeutic efficacy experiments in this specific model. Unlike SOD1 models (where drugs like riluzole, edaravone, or Tofersen for SOD1 ALS have been used as positive controls in some studies), there is currently no universally accepted comparator molecule and/or standard-of-care therapeutic that has shown robust disease modification in the TDP‑43(Q331K) mouse model.
However, several recently published studies have demonstrated that the TDP-43(Q331K) mouse model is pharmacologically responsive to experimental interventions. For example, a pre-print has reported that treatment with dasatinib and quercetin, a senolytic combination, improved motor performance and reduced disease-associated phenotypes in TDP-43Q331K mice (Viteri et al., 2025). In separate studies, M102, a CNS-penetrant small-molecule electrophile that activates the NRF2-ARE and heat-shock response pathways, has also been shown to ameliorate disease-related functional deficits in this mouse model of ALS and FTD (Keerie et al., 2025).
Reach out to our team to discuss study design your therapeutic in the TDP-43Q331K mouse model.
What parts of the TDP-43 pathology does the TDP-43Q331K mouse model recapitulate?
At InnoSer we performed validation experiments to evaluate the disease progression of the mice from 4 weeks until ∼9 months of age, confirming in line with the current literature that the model reproduces several key hallmarks of human TDP-43 proteinopathies.
Progressive motor impairments in the TDP-43 mouse model
- Early phase (4 weeks of age): Compared to TDP-43 WT animals, TDP-43Q331K mice show mild, progressive motor deficits detectable with the inverted grid test. From 7 weeks of age until 9 months of age, mice show reduced CMAP amplitude, indicating early, progressive motor unit dysfunction. The neuronal injury biomarker plasma neurofilament light chain (NfL) is significantly increased from 2 months of age.
- Intermediate phase (~4 months): Robust motor impairments are evident across multiple assays, including rotarod, inverted grid, and weightlifting tests, in line with previously published results (Watkins et al., 2021).
- Late phase (~6 months): Gait abnormalities emerge, consistent with published findings (Watkins et al., 2021), highlighting the progressive nature of motor decline in this model.
Reach out to our team to obtain the full TDP-43 mouse model validation data.
Does the TDP-43Q331K mouse model develop cytoplasmic TDP-43 aggregates?
This TDP-43 mouse model has minimal overexpression of the human TARDBP gene (Watkins et al., 2021; Mitchell et al., 2015); therefore, this single transgenic TDP-43 mouse model shows cytoplasmic TDP-43 accumulation, but no visible cytoplasmic TDP-43 aggregates and nuclear clearing of TDP-43. Consistent with this, cytoplasmic aggregation of TDP-43 into insoluble inclusions in cortical and spinal neurons has been observed in this mouse model at 24 months of age (See figure 5 of Mitchel et al., 2015), making this model more suitable for studying progressive motor deficits and early ALS/FTD-related pathophysiology.
This is in contrast to high TDP-43 overexpression models, such as the double-transgenic TDP-43 mouse model (as described by Mitchell et al., 2015) or the TAR 4/4 TDP-43 mouse model (as described by Wils et al., 2010) where excessive TDP-43 overexpression leads to insoluble TDP-43 cytoplasmic aggregates and nuclear TDP-43 clearing, but early lethality (double transgenic TDP-43 mice live until 8-10 weeks, and similarly TAR 4/4 mice were reported to die at 3-4 weeks of age) limiting the capability to run longitudinal efficacy studies focused on motor function.
What are nerve conduction studies (NCS) and what data do they provide in the TDP-43 mouse model?
The loss of neuromuscular junction innervation and motor neuron death are one of the key hallmarks of human ALS pathophysiology, which are quantitatively captured by nerve conduction studies (NCS). Importantly, in preclinical animal studies, NCS provide you with highly relevant translational data, as NCS are a standard diagnostic and disease monitoring tool in ALS patients.
In mice, electrophysiological recordings are performed on the sciatic nerve, the largest nerve of the peripheral nervous system, supplying the mouse hind limb with both motoric and sensory fiber tracts, using needle electrodes. Compound muscle action potential (CMAP) measures the number and integrity of functional motor units, whereby lower CMAP response indicates fewer functional motor units. Nerve conduction velocity (NCV) measures the speed and nerve signal propagation, serving as a proxy marker for nerve myelination. With nerve damage, white matter is lost, which means it takes longer for the stimulus to reach the connected muscle.
In the TDP-43Q331K mouse model, CMAP amplitude is significantly decreased compared to littermate TDP-43 WT animals already at 7 weeks of age before the onset of robust motor function deficits and remains significantly decreased until at least approx. 9 months of age highlighting the progressive disease phenotype.
What is the translational relevance of including plasma biomarkers such as NfL in my preclinical study package?
Neurofilament light chain (NfL) represents a highly translationally relevant plasma biomarker of neuronal injury in many neurodegenerative diseases, including ALS.
Importantly, the SOD1-targeting antisense oligonucleotide (ASO) Tofersen demonstrated efficacy in reducing plasma and CSF NfL levels in both preclinical and clinical studies. Since then, many ALS clinical trials now routinely incorporate plasma and CSF NfL as a response biomarker to monitor therapeutic effects.
Compared to littermate TDP-43 WT, TDP-43Q331K mice show elevated plasma levels of the neuronal injury biomarker from 5 weeks of age, before onset of robust motor function deficits, indicating the translational relevance of including this biomarker in your preclinical study designs.
What are typical study timelines in the TDP-43Q331K mouse model?
Generally, preclinical efficacy study timelines in the TDP-43Q331K mouse model depend on multiple factors, including your compound’s modality (i.e., is it a small molecule therapeutics, RNA-based therapeutics such as ASOs, siRNA and gene therapy), mechanism of action (i.e., disease-modifying or symptomatic), dosing schedules, route of administration (i.e., ICV, intrathecal, IV via tail vein or retro-orbital), as well as the choice of readouts including motor function battery tests and translational readouts such as nerve conductions studies, and plasma NfL assessments.
Because the TDP-43Q331K model shows early and progressive pathology, compound dosing can typically begin between 2–4 months or 4–6 months of age. Studies usually end around week 8 (∼4 months of age) or week 16 (∼6 months of age), with typical dosing schedules every 2 weeks. Schedules can be adapted based on compound pharmacokinetics, mechanism of action, and study objectives.
How does the SOD1-G93A compare to the TDP-43Q331K mouse model of ALS?
While both SOD1-G93A and TDP-43Q331K models represent well-established preclinical models to study efficacy of novel ALS therapeutics, both differ in terms of target disease, onset and progression of ALS pathophysiology.
Briefly, the SOD1-G93A mouse carries a mutation in the superoxide dismutase 1 (SOD1) gene, which accounts for ~20% of familial ALS cases (5–10% of total ALS). This model reflects SOD1 proteinopathy, with aggregation and misfolding of mutant SOD1 leading to progressive motor neuron degeneration. The TDP-43 model expresses mutant human TDP-43 (i.e., Q331K), a pathology present in ~95% of ALS and ~50% of frontotemporal dementia (FTD) cases. This makes it highly relevant not only for ALS, but also for ALS–FTD spectrum research.
In conclusion, if your therapeutic directly targets SOD1, the SOD1-G93A model is the most suitable. For therapies aiming at broader ALS mechanisms (neurodegeneration, protein aggregation, motor neuron loss, neuroinflammation), TDP-43 models are relevant to a wider patient population.
As a preclinical ALS CRO, InnoSer has expertise in carrying our preclinical studies in both SOD1 and mutant TDP-43 mouse models.
Discover Other Relevant Neurological Disease Models











Discover InnoSer’s Latest Research

InnoSer and Carthera Launch Preclinical Access to the SonoCloud® Ultrasound Platform for CNS Drug Delivery
Researchers developing central nervous system (CNS)-targeted therapies can now leverage Carthera’s SonoCloud® ultrasound-mediated drug delivery platform, helping inform and enhance preclinical development strategies for novel therapeutics. Diepenbeek, Belgium, and...
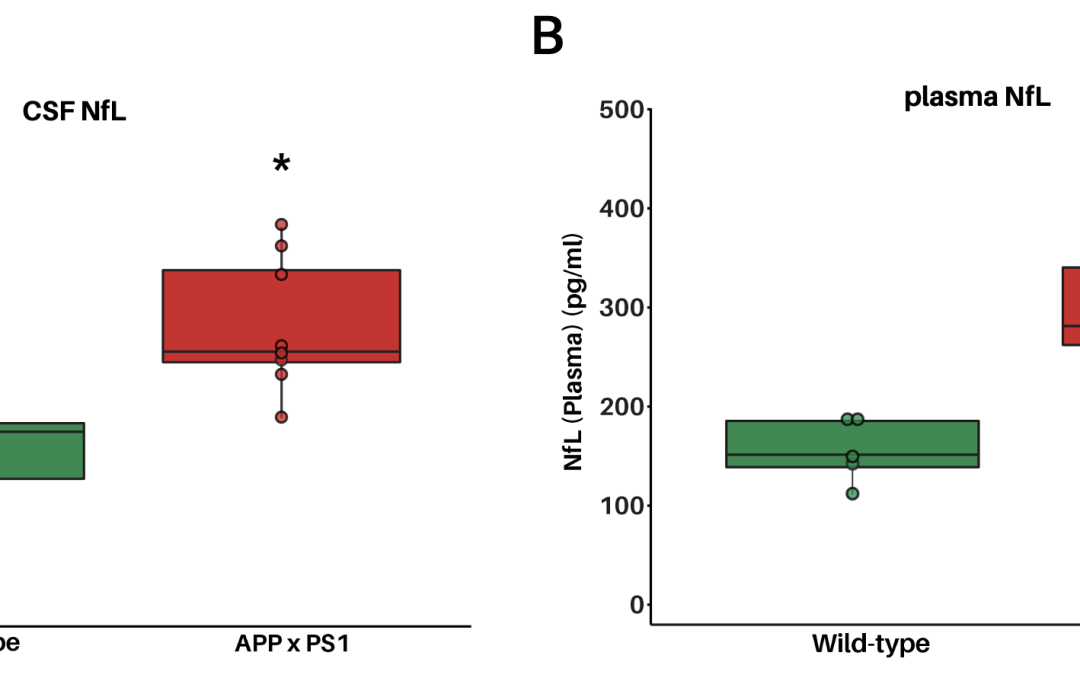
Neurofilament light chain (NfL) in plasma and CSF: A translational biomarker for neurodegeneration in InnoSer’s neurology mouse models
Under normal conditions, neurofilaments, proteins that form the neuronal cytoskeleton, are released at low levels into the extracellular space. From there, they gradually enter the cerebrospinal fluid (CSF) and the bloodstream. Throughout life, levels of...

New insights into TDP-43 mouse model pathophysiology
Earlier this year, we shared data from our phenotyping experiments showing TDP-43 mouse model pathophysiology, consistent with previously published findings in the same mouse model harboring the ALS-linked Q331K mutation in the TDP-43 gene (Watkins et al., 2021). Our...

InnoSer and CureSHANK partner to launch industry-quality preclinical testing service for Phelan-McDermid syndrome (PMS) treatments
Together, CureSHANK, a nonprofit research foundation, and InnoSer, preclinical research CRO, aim to remove barriers to drug development for Phelan-McDermid syndrome treatment by providing the global research community with accessible and industry-standard preclinical...
AAALAC Accreditation
InnoSer has earned the AAALAC accreditation, demonstrating our commitment to responsible animal care and use. AAALAC International is a nonprofit organization that promotes the humane treatment of animals in science through voluntary accreditation and assessment programs. InnoSer’s facilities in the Netherlands and Belgium have been AAALAC-accredited since 2016 and 2020, respectively. Read more about the AAALAC accreditation programme here.
![]()
Animal Welfare
The 3Rs impact everything from policy and regulatory change to the development and uptake of new technologies and approaches. This is why InnoSer has ongoing commitment and monitoring of these processes. The steps we practice maximize our ability to replace, reduce and refine animal involvement and facilitate our commitment to these principles when it comes to research and drug development.
info@innoserlaboratories.com