22q11.2 deletion syndrome (DiGeorge Syndrome) – 22q11 mouse model [Df(h22q11)/+]
Perform preclinical efficacy studies to evaluate your therapeutic compound’s impact on neurodevelopmental and psychiatric phenotypes associated with the 22q11.2 deletion syndrome in humans
22q11 mouse model of DiGeorge Syndrome characteristics
The 22q11 mouse model is an established preclinical research model of 22q11.2 deletion occurring in humans, originally developed and characterized by Didriksen et al., 2017. This deletion, known as DiGeorge syndrome or velocardiofacial syndrome, affects 1 in 2000-4000 individuals, and is associated with a wide range of neurodevelopmental, cardiac, and psychiatric manifestations.
The 22q11 mouse model is frequently utilized to study the underlying disease pathophysiology related to 22q11.2 deletion or evaluate efficacy of therapeutics aimed at treating 22q11.2 deletion syndrome. In humans, most of the microdeletions vary between 3 megabases (Mb) to 1.5Mb in size (accordingly involving approximately 60 or 35 known genes, most of which are expressed in the brain (including TBX1, COMT, DGCR8, PRODH, ZDHHC8). The 22q11.2 deletion mouse model line contains a microdeletion of 1.13Mb spanning from the Dgcr2-Hira on mouse chromosome 16 and encompasses orthologs of all functional genes on the critical human 22q11.2 locus with exception of clathrin heavy polypeptide-like 1 (CLTCL) (Didriksen et al., 2017; Nilsson et al., 2016). This makes the 22q11 mouse model suitable not only for studying 22q11.2 deletion syndrome but also for investigating broader neurodevelopmental, synaptic, and psychiatric phenotypes associated with these genes.
Due to its established link to schizophrenia (Karayiorgou et al., 2010), this model also represents a relevant genetic mouse model of schizophrenia, oftentimes used for evaluating preclinical efficacy of novel therapeutics.
✓ The 22q11 mouse model is suitable for evaluation of novel therapeutics aiming to treat DiGeorge syndrome, schizophrenia as well as other neurodevelopmental and/or psychiatric phenotypes associated with the microdeletion of genes within the 22q11.2 locus
✓ InnoSer’s expert scientific team has confirmed the model’s phenotype including deficits in sensorimotor gating, cognition, and spontaneous behavior, supporting its use in therapeutic efficacy studies
✓ Preclinical efficacy studies using the 22q11 mouse model forms key part of our expertise in rare genetic neurological disorder models, including Fragile X syndrome, vanishing white matter (eIF2B), STXBP1 encephalopathy, Kabuki Syndrome, Phelan-McDermid syndrome, and Angelman syndrome
✓ InnoSer offers comprehensive preclinical support, including biodistribution studies, PK/PD profiling, early-stage toxicology, and tolerability studies

As part of its specialized contract research services across multiple genetic neurological disorders, InnoSer performs efficacy studies using the 22q11 mouse model. In line with initial model characterization studies (Didriksen et al., 2017, Karayiorgou et al., 2010), we observe a robust deficit in sensorimotor gating in 22q11 deletion mice. Building on these findings, we also observed significant impairments in working memory and attention, using automated modified 5CSRT task described by Remmelink et al., (2017), as well as changes in spontaneous behavior assessed with automated PhenoTyper home-cage monitoring, confirming its suitability for preclinical efficacy studies.
Example data featuring the 22q11 deletion mouse model of DiGeorge Syndrome
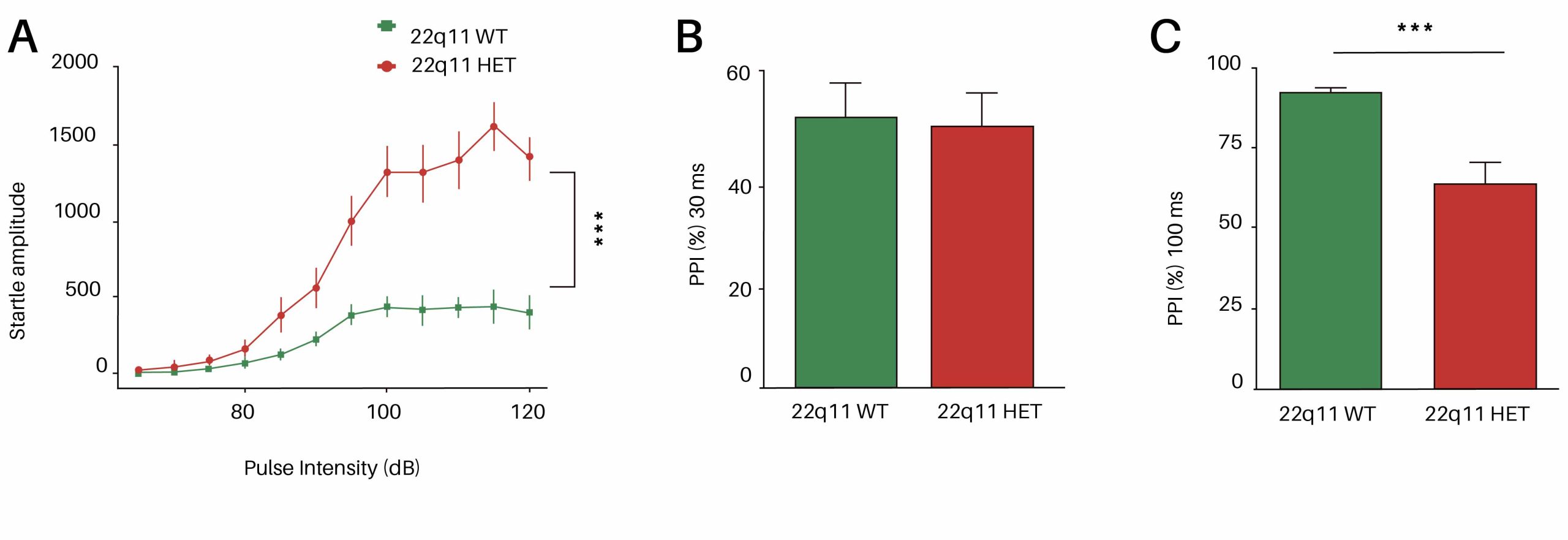
The 22q11 mouse model of human 22q11.2 deletion syndrome show robust deficit in sensorimotor gating as assessed by prepulse inhibition (PPI) test.
(A) Startle amplitude across increasing acoustic intensities (dB) shows that 22q11.2 heterozygous mice display a significantly elevated startle response beginning at 90 dB, indicating heightened baseline reactivity. (B) 22q11.mice show no significant PPI difference at 30 ms (C) but impaired PPI at 100 ms, in line with literature (Didriksen et al., 2017, Karayiorgou et al., 2010).
Key readouts in the 22q11 mouse model of DiGeorge Syndrome
The People Behind Your Research

Thomas Vogels, PhD
Principal Scientist Neurology
Frequently Asked Questions
What diseases or conditions can the 22q11 mouse model be used to study?
The 22q11 mouse model (Df(h22q11)/+) is primarily used to study human 22q11.2 deletion syndrome (also known as DiGeorge syndrome and/or velocardiofacial syndrome), enabling investigation of the underlying neurodevelopmental pathophysiological processes, as well as preclinical efficacy studies evaluating novel therapeutics targeting cognitive and/or behavioral deficits associated with the 22q11.2 microdeletion. In humans, 22q11.2 microdeletions typically range from 3Mb (approx., 60 genes) to 1.5 Mb (approx., 35 genes), known genes that are predominantly expressed in the brain, with examples including DGCR6, PRODH, RANBP1, DGCR8, COMT, TBX1, and HIRA (Maynard et al., 2003).
As described in the original publication (Diederiksen et al., 2017), the mouse model recapitulates the orthologous region of mouse chromosome 16 (MMU 16qA13) corresponding to the human 22q11.2 locus, demonstrating strong construct validity between the model and human 22q11.2 deletion carriers. The Df((h22q11)/+) line carries a 1.13Mb microdeletion spanning Dgcr2-Hira, encompassing orthologs of all functional genes in the critical human 22q11.2 region with except CLTCL, and Igl1 which is not part of human region and has not previously been linked to behavior or cognition in the mouse (Didriksen et al., 2017; Nilsson et al., 2016).
Several genes within the deleted region also contribute to psychiatric vulnerability, making the 22q11 mouse model a relevant platform for studying schizophrenia-relevant phenotypes. Notably, COMT, PRODH, TBX1, and ZDHHC8 are among the most studied schizophrenia risk genes, affecting dopamine signaling, excitatory/inhibitory balance, synaptic stability, and cognitive function. Therefore, the 22q11 mouse model is also suitable to investigate the preclinical efficacy of novel therapeutics targeting schizophrenia-related deficits, such as impairments in sensorimotor gating.
Because the deletion affects multiple conserved genes and pathways, individuals with 22q11.2 deletion syndrome can present with a broad spectrum of phenotypes, including developmental delay, speech and language impairment, intellectual disability, seizures, autism spectrum features, attention deficits, hyperactivity, and behavioral dysregulation, as well as physical manifestations such as facial dysmorphisms, short stature, and hypotonia. Consequently, the 22q11 mouse model may also be applied to study broader neurodevelopmental and psychiatric phenotypes associated with these genes and to evaluate therapeutics targeting downstream circuits, pathways, or specific symptom domains.
Is the 22q11 mouse model suitable for studying the efficacy of therapeutics against schizophrenia?
On the genetic level, the 22q11 mouse model shows strong construct validity for modelling human 22q11.2 deletion syndrome as well as the associated schizophrenia phenotypes, making it a valuable platform for studying schizophrenia-associated pathways and testing the efficacy of preclinical therapeutic candidates. Although 22q11.2 deletion syndrome is one of the known highest genetic risk factors, conferring a staggering 20-fold risk increase for developing schizophrenia (Brzustowicz & Bassett, 2012; Karayiorgou et al., 1995; Xu et al., 2008), schizophrenia remains a complex neuropsychiatric disease in which a combination of genetic, environmental, and epigenetic factors influences the susceptibility to develop the disease.
Accordingly, in humans, the 22q11.2 microdeletion syndrome exhibits incomplete penetrance, and the severity of psychiatric symptoms depends on the interaction between the deletion and the broader genetic background. Therefore, although the 22q11 deletion mouse model captures the genetic risk for schizophrenia along with some key behavioural phenotypes such as sensorimotor gating deficits, you may also consider testing your therapeutics in other, pharmacologically-induced mouse models of schizophrenia (e.g. MK-801 induced mouse model of schizophrenia) to evaluate additional relevant disease mechanisms.
What is the translational relevance of assessing prepulse inhibition (PPI) in the 22q11 mouse model?
In humans, it has been reported that individuals affected by 22q11.2 deletion syndrome show inhibitory impairments such as reduced sensorimotor gating (i.e., filtering out of ‘unnecessary’ stimuli; usually in the form of inhibition of a startle response using the prepulse inhibition [PPI] paradigm), which has also been associated with high-risk schizophrenia symptoms (Karayiorgou et al., 2010).
At InnoSer, we have demonstrated, in line with the literature (Didriksen et al., 2017, Karayiorgou et al., 2010, Stark et al., 2008), that 22q11 mice show a robust deficit in sensorimotor gating assessed by the PPI paradigm. As shown in this figure, PPI at 30 ms interval is not significantly different to WT mice, demonstrating that very fast gating mechanisms are intact; whereas at a longer (100 ms) interval, 22q11 mice show impaired sensorimotor gating, reflecting deficits in higher-order neural circuits (cortical-striatal-pallidal networks) involved in sensorimotor integration.
As sensorimotor gating deficits are also observed in humans, this makes PPI a translationally behavioral endpoint for assessing neural circuit dysfunction and evaluating the efficacy of candidate therapeutics in restoring normal sensorimotor gating in preclinical studies.
Discover Other Relevant Rare Genetic Disease Models















Discover InnoSer’s Latest Research
![Cognitive profiling in the APP[V717I]xTau[P301S] mouse model](https://www.innoserlaboratories.com/wp-content/uploads/2026/07/Figure-1-MWM.png)
Cognitive profiling in the APP[V717I]xTau[P301S] mouse model
In this month's update, we revisit the APP[V717I]xTau[P301S] model with newly in-house generated Morris Water Maze (MWM) data, reconfirming the spatial memory deficits previously described for this model and reinforcing the translational value of the model.The...
![Translational Neuroscience: Comprehensive longitudinal profiling of the Tau[P301S] female vs male mice](https://www.innoserlaboratories.com/wp-content/uploads/2026/06/Female-TauP301S-mice-show-early-spontaneous-hyperactivity-in-automated-home-cages-PhenoTyperTM-229375_1080x323.png)
Translational Neuroscience: Comprehensive longitudinal profiling of the Tau[P301S] female vs male mice
In this month’s newsletter, we highlight and build upon our knowledge working with the Tau[P301S] mouse model (originally described by Allen et al., 2002), and its applicability in preclinical research.Female and male...

Clinically relevant biomarker panels for comprehensive efficacy studies in Alzheimer’s Disease mouse models
InnoSer's validated Alzheimer's disease (AD) mouse models are equipped with translationally relevant biomarker readout panels — spanning amyloid-beta (Aβ) species, phosphorylated Tau isoforms, and the early, sensitive hallmark of neurodegeneration, neurofilament light...
![Proven Compound Susceptible Tau[P301S] Mouse Model for Preclinical Proof of Concept](https://www.innoserlaboratories.com/wp-content/uploads/2026/03/Proven-Compound-susceptible-TauP301S-mouse-model-for-preclinical-proof-of-concept--1080x675.png)
Proven Compound Susceptible Tau[P301S] Mouse Model for Preclinical Proof of Concept
The Tau[P301S] mouse remains one of the most established in vivo models for evaluating therapeutics targeting tauopathies including Alzheimer's disease and frontotemporal dementia (FTD). Presented at AD/PD 2025 in Vienna, this poster provides comprehensive...
AAALAC Accreditation
InnoSer has earned the AAALAC accreditation, demonstrating our commitment to responsible animal care and use. AAALAC International is a nonprofit organization that promotes the humane treatment of animals in science through voluntary accreditation and assessment programs. InnoSer’s facilities in the Netherlands and Belgium have been AAALAC-accredited since 2016 and 2020, respectively. Read more about the AAALAC accreditation programme here.
![]()
Animal Welfare
The 3Rs impact everything from policy and regulatory change to the development and uptake of new technologies and approaches. This is why InnoSer has ongoing commitment and monitoring of these processes. The steps we practice maximize our ability to replace, reduce and refine animal involvement and facilitate our commitment to these principles when it comes to research and drug development.
info@innoserlaboratories.com