22q11.2欠失症候群(ディジョージ症候群) – 22q11マウスモデル [Df(h22q11)/+]
Perform preclinical efficacy studies to evaluate your therapeutic compound’s impact on neurodevelopmental and psychiatric phenotypes associated with the 22q11.2 deletion syndrome in humans
22q1122q11欠失症候群 ディジョージ症候群の特徴を有するマウスモデル
The 22q11 mouse model is an established preclinical research model of 22q11.2 deletion occurring in humans, originally developed and characterized by Didriksen et al., 2017. This deletion, known as DiGeorge syndrome or velocardiofacial syndrome, affects 1 in 2000-4000 individuals, and is associated with a wide range of neurodevelopmental, cardiac, and psychiatric manifestations.
22q11マウスモデルは、22q11.2欠失に関連する基礎的な病態生理学の研究や、22q11.2欠失症候群の治療を目的とした治療薬の有効性評価に頻繁に利用される。 ヒトでは、ほとんどの微小欠失は3メガベース(Mb)から1.5Mbの範囲でサイズが異なり(これに伴い、約60または35の既知の遺伝子が関与し、そのほとんどは脳で発現している(TBX1、COMT、DGCR8、PRODH、ZDHHC8を含む))。 22q11.2欠失マウスモデル系統は、マウス16番染色体上のDgcr2-Hira領域から1.13Mbに及ぶ微小欠失を有し、ヒトの重要な22q11.2遺伝子座上の全機能性遺伝子のオルソログ(クラトリン重鎖ポリペプチド様1 (CLTCL)を除 く、ヒトの重要な22q11.2遺伝子座上の全機能遺伝子のオルソログを含む。この特性により、22q11マウスモデルは22q11.2欠失症候群の研究だけでなく、これらの遺伝子に関連するより広範な神経発達、シナプス、精神疾患の表現型の調査にも適している。
Due to its established link to schizophrenia (Karayiorgou et al., 2010), this model also represents a relevant genetic mouse model of schizophrenia, oftentimes used for evaluating preclinical efficacy of novel therapeutics.
✓ 22q11マウスモデルは、ディジョージ症候群、統合失調症、およびその他の神経発達障害および/または精神疾患の治療を目的とした新規治療法の評価に適している。 その他の神経発達障害および/または精神疾患 表現型 に関連する マイクロ遺伝子欠失に関連する
✓ InnoSerの 専門科学チームは、感覚運動ゲート制御、認知、自発的行動における欠損を含むモデルの表現型を確認した。 行動の障害を含む表現型を確認したを確認し、治療効果研究における本モデルの使用を支持しています
✓ 22q11マウスモデルを用いた前臨床的有効性試験 フォーム 重要な 当社の 専門性の 分野における 希少 遺伝性神経疾患モデルにおける当社の専門知識の一部であり、これには脆弱X症候群、消失性白質(eIF2B)、STXBP1 脳症、歌舞伎症候群、フェラン・マクダーミッド症候群、およびアンジェルマン症候群
✓ InnoSerは、生体内分布試験、PK/PDプロファイリング、初期段階の毒性試験、耐容性試験を含む包括的な前臨床サポートを提供します

複数の遺伝性神経疾患を対象とした専門的な受託研究サービスの一環として、InnoSer社は22q11マウスモデルを用いた有効性試験を実施している。 初期のモデル特性解析研究(Didriksen et al., 2017,Karayiorgou et al., 2010)と同様に、我々は22q11欠失マウスにおいて感覚運動ゲート制御の顕著な障害を観察した。これらの知見に基づき、Remmelink et al. (2017)が報告した自動化修正版5CSRT課題を用いた作業記憶と注意力の著しい障害、ならびに自動化フェノタイピングによる自発行動の変化も確認した。 Remmelink et al., (2017)が報告した自動化修正版5CSRT課題を用いた作業記憶と注意力の著しい障害、ならびに自動化PhenoTyperケージ内モニタリングで評価した自発行動の変化も確認し、前臨床的有効性試験への適性を裏付けています。
Example data featuring the 22q11 deletion mouse model of DiGeorge Syndrome
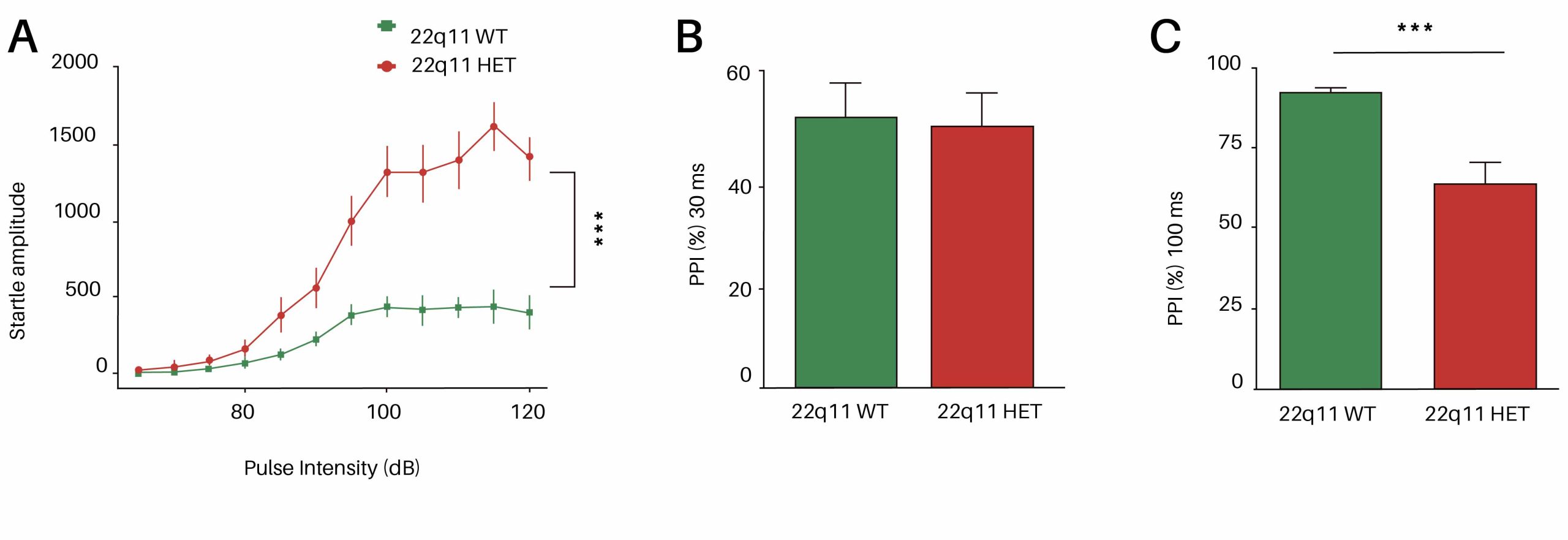
ヒト22q11.2欠失症候群のマウスモデルである22q11マウスは、プレパルス抑制(PPI)試験による評価において、感覚運動ゲート制御に顕著な障害を示す。
(A) 音響強度(dB)の増加に伴う驚愕反応振幅の分析から、22q11.2ヘテロ接合体マウスでは90 dBから有意に高い驚愕反応が示され、 これは ベースライン反応性の増強を示している。 (B) 22q11.miceは30dBにおいて有意なPPI差を示さない ms (C) 100msではPPIが損なわれる msでは 文献(Didriksen et al., 2017, Karayiorgou et al., 2010)。
Key readouts in the 22q11 mouse model of DiGeorge Syndrome
あなたの研究を支える人々

トーマス・フォーゲルス博士
主任神経科学研究員
よくあるご質問
22q11マウスモデルを用いて研究できる疾患や状態にはどのようなものがありますか?
The 22q11 mouse model (Df(h22q11)/+) is primarily used to study human 22q11.2 deletion syndrome (also known as DiGeorge syndrome and/or velocardiofacial syndrome), enabling investigation of the underlying neurodevelopmental pathophysiological processes, as well as preclinical efficacy studies evaluating novel therapeutics targeting cognitive and/or behavioral deficits associated with the 22q11.2 microdeletion. In humans, 22q11.2 microdeletions typically range from 3Mb (approx., 60 genes) to 1.5 Mb (approx., 35 genes), known genes that are predominantly expressed in the brain, with examples including DGCR6, PRODH, RANBP1, DGCR8, COMT, TBX1, and HIRA (Maynard et al., 2003).
As described in the original publication (Diederiksen et al., 2017), the mouse model recapitulates the orthologous region of mouse chromosome 16 (MMU 16qA13) corresponding to the human 22q11.2 locus, demonstrating strong construct validity between the model and human 22q11.2 deletion carriers. The Df((h22q11)/+) line carries a 1.13Mb microdeletion spanning Dgcr2-Hira, encompassing orthologs of all functional genes in the critical human 22q11.2 region with except CLTCL, and Igl1 which is not part of human region and has not previously been linked to behavior or cognition in the mouse (Didriksen et al., 2017; Nilsson et al., 2016).
Several genes within the deleted region also contribute to psychiatric vulnerability, making the 22q11 mouse model a relevant platform for studying schizophrenia-relevant phenotypes. Notably, COMT, PRODH, TBX1, and ZDHHC8 are among the most studied schizophrenia risk genes, affecting dopamine signaling, excitatory/inhibitory balance, synaptic stability, and cognitive function. Therefore, the 22q11 mouse model is also suitable to investigate the preclinical efficacy of novel therapeutics targeting schizophrenia-related deficits, such as impairments in sensorimotor gating.
Because the deletion affects multiple conserved genes and pathways, individuals with 22q11.2 deletion syndrome can present with a broad spectrum of phenotypes, including developmental delay, speech and language impairment, intellectual disability, seizures, autism spectrum features, attention deficits, hyperactivity, and behavioral dysregulation, as well as physical manifestations such as facial dysmorphisms, short stature, and hypotonia. Consequently, the 22q11 mouse model may also be applied to study broader neurodevelopmental and psychiatric phenotypes associated with these genes and to evaluate therapeutics targeting downstream circuits, pathways, or specific symptom domains.
22q11マウスモデルが、22q11.2欠失症候群、統合失調症、および関連する神経発達障害や精神疾患に対する治療候補薬の開発をどのように促進できるか、当社の専門家にご相談ください。
22q11マウスモデルは統合失調症に対する治療薬の有効性を研究するのに適しているか?
On the genetic level, the 22q11 mouse model shows strong construct validity for modelling human 22q11.2 deletion syndrome as well as the associated schizophrenia phenotypes, making it a valuable platform for studying schizophrenia-associated pathways and testing the efficacy of preclinical therapeutic candidates. Although 22q11.2 deletion syndrome is one of the known highest genetic risk factors, conferring a staggering 20-fold risk increase for developing schizophrenia (Brzustowicz & Bassett, 2012; Karayiorgou et al., 1995; Xu et al., 2008), schizophrenia remains a complex neuropsychiatric disease in which a combination of genetic, environmental, and epigenetic factors influences the susceptibility to develop the disease.
Accordingly, in humans, the 22q11.2 microdeletion syndrome exhibits incomplete penetrance, and the severity of psychiatric symptoms depends on the interaction between the deletion and the broader genetic background. Therefore, although the 22q11 deletion mouse model captures the genetic risk for schizophrenia along with some key behavioural phenotypes such as sensorimotor gating deficits, you may also consider testing your therapeutics in other, pharmacologically-induced mouse models of schizophrenia (e.g. MK-801 induced mouse model of schizophrenia) to evaluate additional relevant disease mechanisms.
22q11マウスモデルにおけるプレパルス抑制(PPI)の評価は、臨床応用においてどのような意義を持つのか?
In humans, it has been reported that individuals affected by 22q11.2 deletion syndrome show inhibitory impairments such as reduced sensorimotor gating (i.e., filtering out of ‘unnecessary’ stimuli; usually in the form of inhibition of a startle response using the prepulse inhibition [PPI] paradigm), which has also been associated with high-risk schizophrenia symptoms (Karayiorgou et al., 2010).
At InnoSer, we have demonstrated, in line with the literature (Didriksen et al., 2017, Karayiorgou et al., 2010, Stark et al., 2008), that 22q11 mice show a robust deficit in sensorimotor gating assessed by the PPI paradigm. As shown in this figure, PPI at 30 ms interval is not significantly different to WT mice, demonstrating that very fast gating mechanisms are intact; whereas at a longer (100 ms) interval, 22q11 mice show impaired sensorimotor gating, reflecting deficits in higher-order neural circuits (cortical-striatal-pallidal networks) involved in sensorimotor integration.
As sensorimotor gating deficits are also observed in humans, this makes PPI a translationally behavioral endpoint for assessing neural circuit dysfunction and evaluating the efficacy of candidate therapeutics in restoring normal sensorimotor gating in preclinical studies.
InnoSerの専門家チームに問い合わせて、前臨床データパッケージに患者参画(PPI)を組み込むことの翻訳的価値について詳しく学びましょう。
その他の関連する希少遺伝性疾患モデルを発見する















InnoSerの最新研究を発見する

STXBP1 Foundation and InnoSer validate R122X, a patient-variant mouse model of STXBP1 encephalopathy, to help expand therapeutic opportunities in STXBP1 research
Gene therapies, antisense oligonucleotides (ASOs), RNA editing technologies, and other precision medicine approaches are rapidly transforming the therapeutic landscape for STXBP1 encephalopathy. Currently, around 16 candidate therapies are in development across five...
![Cognitive profiling in the APP[V717I]xTau[P301S] mouse model](https://www.innoserlaboratories.com/wp-content/uploads/2026/07/Figure-1-MWM.png)
Cognitive profiling in the APP[V717I]xTau[P301S] mouse model
In this month's update, we revisit the APP[V717I]xTau[P301S] model with newly in-house generated Morris Water Maze (MWM) data, reconfirming the spatial memory deficits previously described for this model and reinforcing the translational value of the model.The...
![トランスレーショナル神経科学:Tau[P301S]を保有する雌マウスと雄マウスの包括的な縦断的プロファイリング](https://www.innoserlaboratories.com/wp-content/uploads/2026/06/Female-TauP301S-mice-show-early-spontaneous-hyperactivity-in-automated-home-cages-PhenoTyperTM-229375_1080x323.png)
トランスレーショナル神経科学:Tau[P301S]を保有する雌マウスと雄マウスの包括的な縦断的プロファイリング
今月のニュースレターでは、Tau[P301S]マウスモデル(Allenら、2002年に初めて報告された)を用いた研究から得られた知見に焦点を当て、その前臨床研究における応用可能性について詳しく解説します。雌と雄……

アルツハイマー病マウスモデルにおける包括的な有効性研究のための臨床的に意義のあるバイオマーカーパネル
InnoSer社の検証済みアルツハイマー病(AD)マウスモデルには、トランスレーショナル研究に関連するバイオマーカー測定パネルが備わっています。これには、アミロイドベータ(Aβ)種、リン酸化タウアイソフォーム、そして神経変性の早期かつ高感度の指標であるニューロフィラメント軽鎖などが含まれます...
AAALAC認定
InnoSerはAAALAC認証を取得し、責任ある動物ケアと利用への取り組みを実証しています。AAALAC Internationalは、自主的な認証および評価プログラムを通じて科学における動物の適切な扱いを推進する非営利組織です。InnoSerのオランダおよびベルギー施設は、それぞれ2016年および2020年よりAAALAC認証を取得しています。AAALAC認証プログラムの詳細はこちらをご覧ください。
![]()
動物福祉
3R原則は、政策や規制の変更から新技術・手法の開発と普及に至るまで、あらゆる分野に影響を及ぼします。このためInnoSerは、これらのプロセスに対する継続的な取り組みと監視を実施しています。当社が実践する手順は、動物実験の代替・削減・改善を最大限に実現し、研究および医薬品開発におけるこれらの原則への取り組みを促進します。
info@innoserlaboratories.com